Making Pharma AI-Ready: Applying the FDA's Draft Guidance
July 16, 2025
Back in January, the FDA released draft guidance for AI: Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products. While this guidance could change based on public feedback and FDA review, it marks an important step forward in defining how AI can be used across the drug development lifecycle.
The guidance lays out a structured framework for when and how to integrate AI in submissions. In doing so, the FDA is signaling a shift: AI now has a regulatory pathway to reduce or replace physical validation runs, manufacturing investigations, and even clinical trial activities.
This article highlights the key points of the FDA’s draft guidance and explores how they might apply to two high-impact use cases in process development and manufacturing.
While this is only draft guidance, the message is clear: the regulatory path is taking shape, empowering biopharma companies to embrace AI with greater confidence.
What the FDA’s guidance means and how to make it actionable
The FDA’s draft guidance clarifies when and how AI models can be used to support regulatory decision-making across the drug lifecycle. It outlines expectations for models that inform decisions related to drug safety, effectiveness, or quality, and draws a boundary around use-cases that fall outside regulatory scope, such as drug discovery or internal operations.
The 7-step evaluation framework
At the heart of the FDA’s guidance is a 7-step framework for evaluating whether an AI model is credible for its intended use-case. The FDA is not prescribing how to develop AI models, but rather offering a structured way to assess and address model risk. The framework helps sponsors determine what level of validation is appropriate and how to prepare their documentation.
To help scientists better understand the guidance, we’ve given more detail on what each of the 7 steps entails, below:

Evidence Requirements Scale with Risk
The FDA expects that the depth and rigor of evidence matches the potential risk of the AI model:
- Low-risk uses (e.g., supporting non-critical decisions): May require only basic documentation, a description of model design, and limited performance metrics.
- High-risk uses (e.g., informing pivotal decisions or justifying acceptance criteria): Require a more detailed credibility assessment, including model validation, verification, performance uncertainty estimates, and lifecycle monitoring plans.
Sponsors are expected to summarize their evidence in a credibility assessment report. This is a document that records the model’s purpose, design, risk classification, validation activities, performance results, and any deviations from the planned assessment. It should help demonstrate the model’s fitness for its intended regulatory use and may be shared with the FDA in submissions, during formal meetings, or kept on file for inspection, depending on the context.
Engage early and often
A central message from the draft guidance is the value of early and ongoing engagement. Sponsors are encouraged to check in before and during model development, especially when defining the model’s intended role and risk level (Steps 1–3) and planning credibility activities (Step 4).
These early conversations are also an opportunity to align on lifecycle management, or how the model will be monitored and maintained after deployment, since AI model performance can change with new data and other model updates.
When the Framework Applies - and When It Doesn’t
This framework applies when an AI model is used to generate information or data that directly supports regulatory decision-making related to a drug’s safety, effectiveness, or quality. In these cases, a structured credibility assessment is expected.
On the other hand, AI models used outside of regulatory decision-making, such as in drug discovery or to support internal efficiencies, generally do not fall under the framework.
Examples of out-of-scope use-cases include:
- Designing more efficient experiments
- Identifying potential process risks for internal mitigation
- Drafting regulatory documents or organizing submission content
In borderline cases, the FDA encourages early engagement to determine whether the proposed AI use falls within the scope.
Illustrative Use Cases: Applying theFDA's 7-Step AI Guidance
One of the most compelling applications of AI in biopharma is its potential to enable simulation to replace physical experiments and to enable better process understanding on the fly. This shift can dramatically accelerate time to market, reduce development and manufacturing costs, and enhance process robustness. But what does that look like in practice, and how might regulators evaluate the credibility of such an approach?
To demonstrate how the FDA’s 7-step AI guidance can be applied in practice, we explore two medium-to-high risk use cases: one in Process Development, the other in Manufacturing. Both involve medium/high risk scenarios with AI contributing to decisions impacting product quality.
Using AI to determine CPPs in Process Development
To illustrate this, consider how a biopharmaceutical company would use the FDA’s 7-step risk-based framework to support a simulation-driven control strategy during late-stage upstream process development.
In this scenario, the company is finalizing its control strategy for a new continuous monoclonal antibody (mAb) process, focusing on optimizing the perfusion rate. The perfusion rate is a critical process parameter (CPP) that affects nutrient exchange, metabolite buildup, cell viability, and ultimately product titer and quality attributes like glycosylation and charge variants.
Traditionally, the process development team would have executed several bioreactor runs at different perfusion rates (e.g., 1.0, 1.5, and 2.0 RV/day) to compare outcomes.
Instead, the team deploys a hybrid AI model that combines a mechanistic bioprocess simulator with a machine learning layer trained on historical run data. The model simulates process performance across a range of perfusion rates and identifies 1.75 RV/day as the optimal setpoint. To confirm model predictions, the team conducts an experimental validation run at 1.75 RV/day, measuring critical quality attributes (CQAs) to ensure predicted and measured product quality attributes match before including the CPP in their commercial filing.
Because the AI model output informs a filed CPP, the company uses the FDA’s 7-step framework to assess and document the model’s credibility for its intended regulatory use. This includes defining the model’s role and context of use, assessing model risk, and developing a credibility assessment plan that outlines evidentiary sources (e.g., historical bioreactor data, product quality testing, prior development studies) and performance criteria (e.g., prediction accuracy, agreement between simulated and observed data, confidence intervals).

It’s also possible the FDA could suggest changes during review, like lowering model influence by adding other evidence sources or tightening performance criteria.
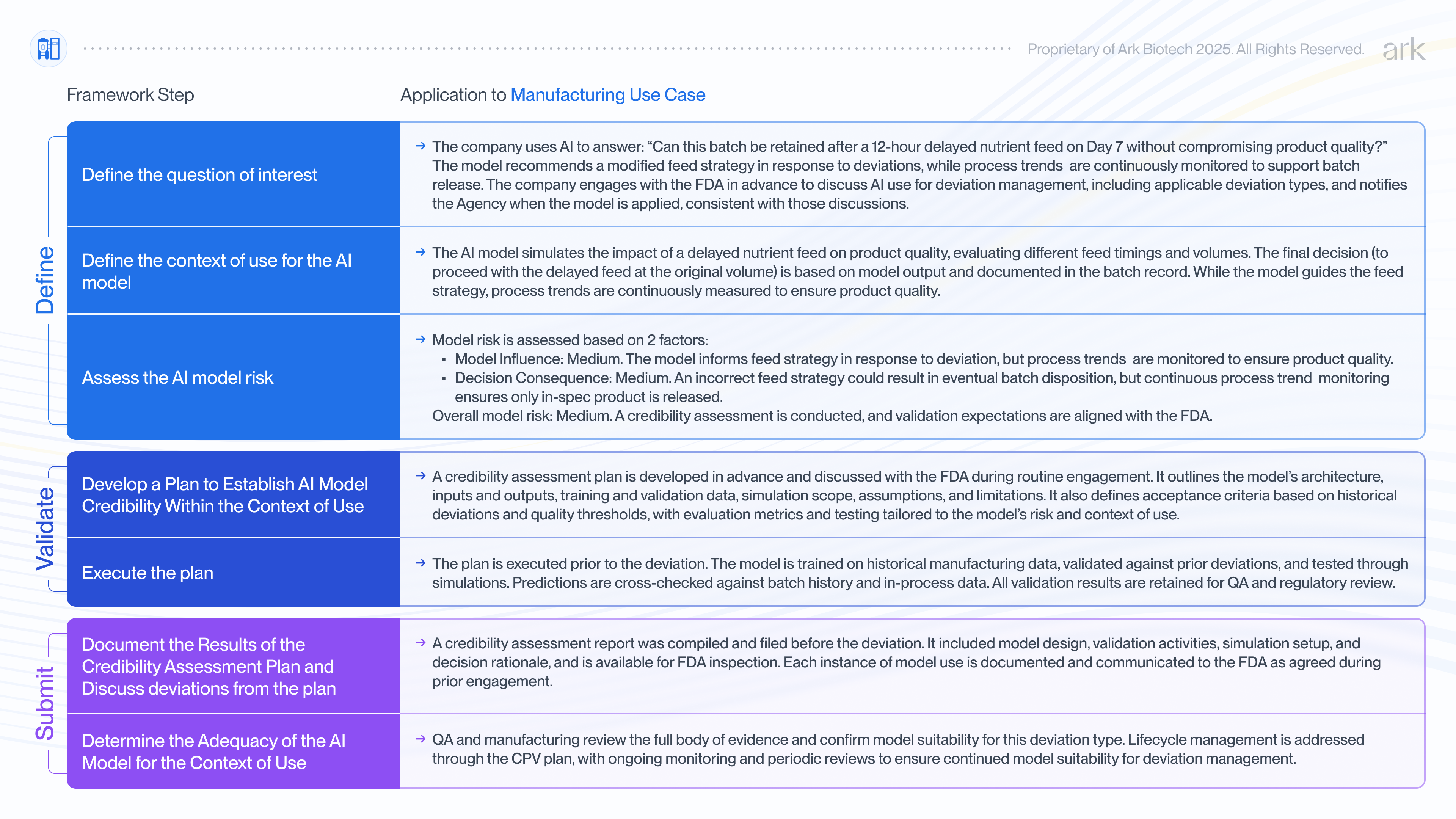
Using AI to assess a deviation in commercial manufacturing
AI can also play a critical role in manufacturing, where deviations can trigger time-consuming root cause investigations or unnecessary batch rejection. In this scenario, we consider how a biopharmaceutical company might apply the FDA’s 7-step framework to assess and document the credibility of an AI model used to guide feed strategy decisions in response to a deviation during commercial production of a therapeutic protein.
An equipment malfunction in the water-for-injection (WFI) system delays preparation of the nutrient feed in a fed-batch process. The feed is normally administered on specific days within a ±4-hour window, but due to the malfunction, it will be 12 hours late. The manufacturing and quality teams must quickly assess whether this delay impacts product quality and whether adjustments to feed timing or volume are needed.
Traditionally, the team would search process characterization data for precedent or conduct investigatory experiments. But such data typically lacks coverage of specific deviation scenarios and targeted experiments take time to conduct. In the absence of strong supporting evidence and a need for a quick decision, the default is often to discard the batch.
Instead, the company deploys a hybrid AI model, similar to the example above, trained on commercial manufacturing data and maintained under the Continued Process Verification (CPV) plan. The model simulates the impact of various feed timings and volumes. It predicts that feeding 12 hours late, without changing the volume, will keep all critical quality attributes (including glycosylation, charge variants, aggregates, and purity) within specification.
The team proceeds with the delayed feed at the original volume and continues to closely monitor process trends throughout the production run. This real-time monitoring provides assurance that the process behaves as expected and the product remains within quality limits ensuring it is safe for release.
The decision and supporting rationale are documented by Quality Assurance in the deviation report and batch record, both of which are subject to FDA inspection. Data from this batch are included and used to update the model under the CPV plan, establishing a precedent to strengthen the model.
Prior to the deviation occurring, the company had engaged with the FDA to discuss model use for deviation management and response, including the types of scenarios the model may address. This process involved the company applying the FDA’s 7-step framework to assess and document the model’s credibility. As specific deviations occur, the company notifies the FDA when the model is applied, consistent with those prior discussions.
A Regulatory Path to Faster, Safer, and Smarter Drug Development
The FDA’s draft guidance is an important step toward making AI a credible, regulatory-grade tool in drug development. By outlining how AI models can support decisions in regulatory submissions, the agency acknowledges both the risks and the potential of these technologies. With the right guardrails in place, AI can be safely integrated into development workflows, opening the door to faster, more efficient, and more informed decision-making.
The examples in this article show how AI can support specific, high-impact decisions. But the real long-term value of AI extends beyond efficiency gains. It lies in enabling a safer, more reliable approach to drug development. By using AI to explore the full design space and assess process risks across a wide range of operating conditions, teams gain a deeper understanding of process behavior. This enhanced understanding empowers them to develop more robust control strategies, proactively anticipate potential failure modes, and iterate more confidently, ultimately improving product safety and quality.
At Ark, we’re building AI-powered bioreactor simulation software to make this future a reality. By grounding process decisions in deeper insight, our goal isn’t just to help teams move faster or reduce costs, it’s to enable more life-saving therapies to reach patients sooner.


